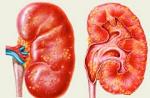
Sindromul Prader-Willi este o tulburare genetică diagnosticată cu o frecvență de 1: 10.000 până la 1: 25.000 de cazuri. Mai mult, numărul de persoane cu această tulburare diferă nu numai în funcție de prevalența bolii în diferite regiuni, dar, de asemenea, se corelează direct cu dezvoltarea sănătate... În absența geneticii în clinică, simptomele șterse, părinții și alții cred adesea că copilul pur și simplu „mănâncă bine”, nu îi plac jocurile active și este ușor întârziat în dezvoltare, poate are nevoie de mai multă atenție și de activități suplimentare. Și până la intrarea în școală, se dovedește că excesul de greutate și întârziere dezvoltare mentală nu a condus factori sociali, dar o patologie genetică, a cărei corecție a trebuit să înceapă chiar de la naștere.

Boala poartă numele cercetătorilor care au descris pentru prima dată complexul simptomelor. Pediatrii elvețieni Andrea Prader și Heinrich Willie au selectat un grup de copii cu dizabilități de dezvoltare similare din masa pacienților tineri și au descris-o la mijlocul secolului trecut drept o boală separată.
Ulterior s-a dezvăluit că apariția acestui sindrom este cauzată de absența sau disfuncționalitatea a 7 gene (segmentul 11.2-q13) pe cromozomul 15, iar datorită diferenței de simptome în absența sau încălcarea parțială, severitatea bolii poate varia semnificativ - de la cazurile grave diagnosticate la naștere, la forme șterse.
Cauza inițială a încălcărilor în genom nu este clară, dar această patologie este întotdeauna moștenită prin linia masculină în prezența unei ideosomii genetice adecvate în materie și este transmisă de la tată la copii, deși copii sănătoase ale genelor pot duce la moștenire ascunsă, iar copiii cu o astfel de boală se nasc în persoane fără sindrom.
Predispoziția genetică este un factor suplimentar care crește vârsta diagnosticării la copii înainte de examinarea preșcolară (și chiar mai târziu): manifestări externe Sindromul Prader-Willi, care este destul de necaracteristic pentru orice altă familie, cu o moștenire genetică evidentă, arată foarte logic: copilul arată doar ca tatăl său, atât ca aspect, cât și cu apetit și caracter.
Comentariu de expert
Sindromul Prader-Willi - suficient patologie rară dezvoltarea, însă, este periculoasă nu numai prin manifestările sale, ci și prin necunoscut. Aspectul caracteristic al copiilor, tulburări de comportament, întârzierile de dezvoltare pot însoți nu numai multe alte boli, dar pot fi moștenite de la părinți. Cu o astfel de imagine, este clar că contactul specialiștilor poate să trage mai mult pe întreaga perioadă preșcolară, iar uneori copiii rămân fără diagnostic corect și tratament necesar, pur și simplu pentru că sunt foarte asemănătoare cu tata, iar aceasta este considerată norma.
În practica mea, a existat un băiat pe nume Volodya, care a fost adus la un centru de dezvoltare la vârsta de 8 ani, cu intenția de „a se pregăti pentru școală”. Copilul nu a trecut testele preșcolare în primăvară din cauza inconsistenței nivelului de dezvoltare (memorie, atenție, procese de gândire) cu cerințele unei instituții de învățământ general, dar părinții au crezut că a fost vina că Vova nu a participat la grădiniță, ci a petrecut timp în principal cu o bunică bătrână din sat. ... De asemenea, ea l-a „hrănit” lapte de capra, greutate excesiva băiatul avea destul.
Testele de dezvoltare Volodya au „eșuat” într-adevăr, în funcție de vârstă, au diferit poftă bună, excelentă amintire vizuală și un poftă pentru jocul constructorilor Lego, uneori era încăpățânat și „explodat”. În general, acesta corespundea nivelului unui copil de cinci ani. Pentru destul de mult timp, la aproximativ trei luni de la începerea cursurilor, specialiștii noștri au considerat, de asemenea, că motivul problemelor băiatului a fost neglijarea socială și pedagogică. Dar timpul a trecut, scrisorile nu au mai fost amintite, iar tatăl a venit să-l ia pe Volodya cu simptome externe și mai pronunțate: statură scurtă, plinătate, podul pronunțat al nasului, ochi în formă de migdale, acromicria etc. Din acel moment imaginea a început să se clarifice.
După lungi negocieri cu mama sa și convingerea tatălui său, Vova a primit în sfârșit o consultație de la un medic genetician și o sesizare la o școală specială. A devenit clar că școala generală de învățământ nu era încă pregătită să învețe copilul, nu exista o clasă de corecție și s-au pierdut bani pentru studierea grundului într-un centru privat din acest moment nu are sens, avem nevoie de kinetoterapeuți-defectologi, endocrinolog, dieta speciala și multă muncă pentru a compensa timpul pierdut și pentru a preveni comorbiditățile.
Intelectul, care suferă la pacienții cu sindrom Prader-Willi, în cazul moștenirii familiei a bolii, complică corectarea. Adesea, tatăl nu vrea să accepte că el însuși este bolnav, iar copilul are nevoie de tratament. Schimbările frecvente de dispoziție, rigiditatea crescută, agresivitatea, care sunt, de asemenea, simptome ale bolii, duc la necesitatea unei lucrări preliminare îndelungate cu membrii familiei înainte de a începe cursurile directe cu copiii. De asemenea, cuplurile au nevoie de consiliere genetică înainte de a concepe alți copii.

Probabilitatea dezvoltării patologiei poate fi suspectată chiar și în procesul de gestație. Este necesară consultarea și examinarea cu un medic genetician scăderea activității făt, este poziție greșită în cavitatea uterină, polihidramnios, precum și niveluri inadecvate gonadotropină corionică o persoană dintr-o femeie însărcinată.
Sindromul Prader-Willi este o afecțiune genetică rară. Dezvoltarea bolii este asociată cu următoarele procese:
- disfuncția genelor;
- lipsa de exprimare
Principala funcție a genelor este de a activa funcția sistemului genetic. Manifestările la bărbați sunt lipsa părului facial.
Pubertatea este însoțită de lipsa părului. Descrierea patologiei se încadrează în anii cincizeci. Acest sindrom este observat relativ rar.
Patologia a fost descrisă de mulți oameni de știință.
Sindromul Prader-Willi - etiologie
Caracteristica patologiei:
- lipsa a șapte gene;
- proces de exprimare
Genele sunt transmise de la tată. Cu patologii cromozomiale ale mamei, a. Boala se manifestă prin absența unei copii a genelor.
Sindromul Prader-Willi - semne
Simptomele sunt următoarele manifestari clinice:
- proces de displazie articulară;
- prezența hipotoniei;
- supraponderal;
- scăderea coordonării mișcării;
- scăderea densității osoase;
- disponibilitate mărime mică picioare;
- prezența unei dimensiuni mici de perie;
- creștere scăzută;
- stare de somn;
- ochi alungiți;
- coloana vertebrală este curbată
Semne externe:
- consistența groasă a salivei;
- scăderea funcției sexuale;
- încălcarea dezvoltării dinților;
- infertilitate
Alte semne:
- probleme mentale;
- tulburări de vorbire;
- încălcarea pubertății;
- scăderea funcției motorii
Alte manifestări la pacienți:
- dimensiunea podului nasului este mare;
- prezența unei frunți înguste;
- prezența unei frunți înalte;
- prezența unei buze înguste
Sindromul Prader-Willi - metode de diagnostic
Diagnosticul se efectuează în pântece. Semne de leziuni intrauterine:
- mobilitatea fetală este afectată;
- poziția copilului este încălcată;
- prezența polihidramniosului
Diagnosticele includ cercetare genetică... Grup de risc - un nou-născut cu o scădere tonusului muscular... Boli similare:
- semne ale sindromului Down;
- miopatie
Simptome tardive:
- secțiune cezariană are semne;
- prezentare fesier;
- scăderea funcției de supt;
- slabiciune musculara;
- tulburare respiratorie;
- prezența hipogonadismului
Asemănarea semnelor externe ale copiilor între ei.
Asemănări între sindromul Angelman și Prader-Willi
Sindroamele patologice sunt similare. Mutația lor se unește. O enzimă este un produs genic.

Boala este similară cu numele unui medic pediatru.
Sindromul Prader-Willi - terapie
Originea congenitală a bolii. Nu au fost studiate terapiile. Care sunt metodele de terapie?
Exista procedura medicala... Metode de tratament:
- masaj;
- tratament specific;
- defectolog;
- kinetoterapeut
Terapia medicamentoasă include:
- utilizarea hormonului de creștere;
- gonadotropine
Manifestări ale semnelor în câmpul masculin:
- scăderea gonadelor;
- micropenis;
- semne de criptorhidie
În absența prolapsului testicular, se folosește intervenția chirurgicală. Terapia cu hormoni este de asemenea folosită. Alimentație dietetică include următoarele:
- scăderea glucidelor;
- scaderea grasimilor
Controlul este legat de nutriție. Apetitul pacienților nu este deranjat. complicaţii:
- probleme cu respirația noaptea;
- semne
Sindromul Prader-Willi - prognostic
Poate dezvoltarea sindromului la nașterea secundară. Motivul este eșecul genic. Riscurile sunt reduse la ștergerea genelor. 50% risc pentru daune mutaționale.

Dar metodă importantă prevenirea este testarea genetică. Semnele care sunt salvate:
- dezvoltarea vorbirii este întârziată;
- dezvoltarea psihică retardată
Întârzierile de dezvoltare pot fi următoarele:
- personaj intens;
- natura medie a rezultatului;
- caracter slab întârziat
Mai des, diagnosticul mediu de informații este diagnosticat la astfel de pacienți. Acești copii au memorie bună. Are un caracter de lungă durată.
Copiii au următoarele proprietăți:
- capacitatea de a citi;
- vocabular pasiv;
- înțelegerea vorbirii nu este afectată;
- memoria auditivă afectată;
- abilități medii matematice
Simptomele bolii:
- apetit crescut;
- creșterea ghrelinei de hormoni
Aceste semne pot indica o scădere a numărului de celule din hipotalamus. Mai rar, aceste semne nu sunt asociate cu acest lucru.
Durata de viata
Cu această boală, speranța de viață scade cu tulburări respiratorii... Dar, cu diagnosticul și terapia în timp util, calitatea vieții este îmbunătățită. Speranța de viață în în acest caz va depinde de următoarele:
- tratament corect;
- prezența unui nivel sever sau moderat de deteriorare;
- simptome de detresă respiratorie
O persoană poate muri în vis când acest sindrom... Motivul este insuficiența respiratorie, sufocarea. Dacă patologia este caracterizată de semne externe, atunci utilizați interventie chirurgicala.
Este operația care prelungește viața unei persoane, îmbunătățește calitatea acesteia. Dupa toate acestea semne exterioare avea mare importanță pe parcursul bolii. Dar terapia trebuie efectuată sub supravegherea unui medic.
![]()
Descriere:
Sindromul Prader-Willi este o anomalie genetică rară. În sindromul Prader-Willi, aproximativ 7 gene de la al 15-lea cromozom moștenit de la tată sunt absente sau nu sunt exprimate.
Kariotip 46 XX sau XY, 15q-11-13. Boala a fost descrisă pentru prima dată de pediatrii elvețieni A. Prader și H. Willi în 1956.
Conform registrului asociațiilor de pacienți cu sindrom Prader-Willi, în Statele Unite și Canada, în decembrie 1986, erau 1595 de pacienți. ÎN anul trecut a fost posibil să se stabilească frecvența populației patologiei, care este de 1: 10.000 - 1: 20.000.
Cauzele sindromului Prader-Willi:
Autorii, care au descris pentru prima dată sindromul, au sugerat un mod autosomal recesiv de moștenire a bolii. Apoi au existat rapoarte despre posibilitatea transmiterii autosomale dominante a bolii. Cazurile de patologie familiale observate ar putea servi drept confirmare a acestor ipoteze. Cu toate acestea, majoritatea descrise observații clinice Sindromul Prader-Willie a fost sporadic.
Studiile ulterioare au stabilit anumite anomalii cromozomiale la copiii cu sindromul Prader-Willi. Analiza citogenetică a arătat că anomalii cromozomiale la pacienți au fost reprezentate fie translocări (t 15/15), fie mozaicism. În 1987, au apărut primele rapoarte despre microdeletia cromozomului 15. Cu toate acestea, identificarea finală a modificărilor cromozomiale în sindromul Prader-Willi a devenit posibilă numai după introducerea în practică a metodelor de cercetare genetică moleculară.
S-a stabilit acum că dezvoltarea sindromului Prader-Willi este asociată cu deteriorarea regiunii critice a cromozomului 15 (segmentul q11.2-q13). În același timp, s-a dovedit că deteriorarea aceleiași regiuni a cromozomului 15 este observată într-o altă boală - sindromul Angelman, tablou clinic care diferă semnificativ de sindromul Prader-Willi și se caracterizează printr-o decelerare timpurie (la vârsta de 6-12 luni) dezvoltarea psihomotorie, microcefalie, tulburări de vorbire (în 100% din cazuri), ataxie, râs violent necontrolat, convulsii epileptiforme frecvente, expresii faciale specifice.
Astfel, în ciuda afectării aceluiași locus al cromozomului 15 în sindroamele Prader-Willi și Angelman, manifestările clinice ale ambelor boli sunt puternic opuse.
O explicație a diferențelor fenotipice a fost obținută doar în ultimii ani. S-a dovedit că dezvoltarea acestor boli este asociată cu noi fenomene genetice - amprenta genomică și disomia uniparentală.
Amprenta genomică este un fenomen nou descoperit datorită progreselor în genetica moleculară. Înseamnă o expresie diferită a materialului genetic (alele omologe) în cromozomi în funcție de originea paternă sau maternă, adică. indică influența părinților asupra fenotipului copilului. Până acum, se credea că contribuția la manifestarea (expresia) genelor tatălui și mamei este echivalentă.
De fapt, imprimarea genomică este un modificator complex sexual și dependent de țesuturi al activității genice a unor loci cromozomici, în funcție de originea parentală a acestora. Manifestări de imprimare genomică au fost identificate și în alte boli - Sotos, Beckwith-Wiedemann, sindroamele Silver-Russell, fibroza chistică și altele.
Disomia uniparentală (uniparentală) este moștenirea ambelor cromozomi de la doar unul dintre părinți. Timp de mulți ani, se credea că o astfel de moștenire era imposibilă. Doar cu ajutorul markerilor genetici moleculari a fost posibil să se demonstreze posibilitatea disomiei uniparentale. Natura disomiei uniparentale nu a fost pe deplin elucidată, dar s-a stabilit că își datorează originea unei serii de tulburări genetice și biochimice.
Trebuie menționat că este imposibilă detectarea microdeletiei sau disomiei uniparentale folosind un studiu convențional al compoziției cromozomiale a cariotipului. Pentru aceasta, se folosesc metode genetice speciale citogenetice și moleculare - analiza prometafazei, utilizarea markerilor ADN din anumite regiuni ale cromozomului 15 (studiul proceselor de metilare) etc.
Astăzi, sindroamele Prader-Willi și Angelman servesc ca un model general acceptat pentru studierea fenomenelor noi și complexe în genetica clinică - amprenta genomică și disomia uniparentală.
S-a stabilit că sindromul Prader-Willi poate fi cauzat de două mecanisme principale. Prima dintre ele este microdeletia cromozomului 15 (15q11.2-q13), care este întotdeauna de origine paternă. Al doilea este izodisomia maternă, adică. când ambii cromozomi 15 sunt de la mamă. Dezvoltarea sindromului Angelman, dimpotrivă, este asociată cu o microdeletie din aceeași regiune a cromozomului 15, dar de origine maternă sau izodisomie paternă. Cele mai multe (aproximativ 70%) cazuri de sindrom Prader-Willi se datorează microdelecțiilor, restul se datorează disomiei. În același timp, este de remarcat absența diferențelor clinice între pacienții cu microdeletie și izodisomie.
Patogenie:
Patogenia sindromului Prader-Willi rămâne până acum slab înțeleasă. Se sugerează că la pacienți se datorează creșterii semnificative (de peste 10 ori) a sintezei grăsimii din procese de acetat și a lipolizei extrem de scăzute.
tipul hipogonadotrop poate fi asociat cu disfuncția hipotalamusului, în principal în zona nucleelor \u200b\u200bventromediale și ventrolaterale. Corectitudinea acestui punct de vedere este confirmată de eficacitatea tratamentului pacienților produse farmaceutice (clomifen), ceea ce duce la creșterea nivelului plasmatic al hormonului luteinizant, testosteronului, normalizarea excreției renale a gonadotropinelor, spermatogenezei și apariția caracteristicilor sexuale secundare.
Una dintre explicațiile pentru hipopigmentarea pielii, părului și irisului este o scădere a activității tirozinazei în foliculi de păr și melanocite, precum și o scădere a pigmentului în retină.
Se atrage atenția risc crescut dezvoltarea leucemiei la pacienții cu sindrom Prader-Willi. Studiile au arătat o scădere a reparației ADN-ului (până la 65% față de 97% în copil sanatos) în limfocitele pacienților cu această patologie. Este posibil ca capacitatea scăzută de reparare a ADN-ului să joace un rol fatal în dezvoltarea neoplasme maligne la persoanele cu sindrom Prader-Willi.
Simptomele sindromului Prader-Willi:
Bebelușii cu sindrom Prader-Willi sunt de obicei născuți pe termen complet, cu puțini hipotrofie intrauterină și adesea în. Prezentarea Breech este observată în 10-40% din cazuri.
Pe parcursul bolii se pot distinge două faze: prima este caracteristică copiilor cu vârsta cuprinsă între 12-18 luni. Se caracterizează prin hipotonie musculară severă, reflexe reduse - Moro, supt și înghițire, ceea ce face dificilă hrănirea copilului. Al doilea vine mai târziu, după câteva săptămâni sau luni. Apărea, senzație constantă foamea, ceea ce duce la dezvoltarea obezității, iar depunerea de grăsime este observată mai ales pe trunchi și în extremitățile proximale.
Hipotensiunea musculară scade treptat și până la vârsta școlară dispare aproape complet. Picioarele și mâinile pacienților sunt disproporționat de mici - acromicria. La copii se remarcă hipogonadismul (la băieți, hipoplazia penisului, scrot și la fete, subdezvoltarea labiilor și, în 50% din cazuri, a uterului).
Creșterea pacienților este adesea redusă. La 75% dintre copii se observă hipopigmentarea pielii, părului și irisului. Este adesea diagnosticat. Dezvoltarea psihomotorie rămâne în urma normei de vârstă - IQ-ul este de la 20 la 80 de unități. (la o rată de 85-115 unități). Vorbirea este dificilă, vocabularul este redus. Pacienții sunt binevoitori, starea de spirit este caracterizată de schimbare frecventă... Sunt descrise tulburările de coordonare, strabismul.
Există și alte anomalii: microdontia, hipoplazia cartilajelor atrii, ectropion (eversiunea secolului),.
Sindromul Prader-Willi - destul de rar starea patologică, care este moștenit. Se caracterizează prin faptul că șapte gene de pe al cincisprezecelea cromozom patern nu funcționează (sau nu funcționează pe deplin).
Diagnosticul apare la un copil la 15-25 de mii. Caracteristicile sexuale nu au niciun efect, deoarece procesul patologic este observat la nou-născuți, atât la bărbați, cât și la femei. Rasa, naționalitatea nu afectează cât de des este diagnosticat problema.
Un semn specific al unui proces patologic la copiii sub un an este. A relevat un fenomen anormal similar în raport cu sindromul Prader-Willi - sindromul Angelman. În timpul apariției acestui tip de patologie, nu sunt paterne, ci cromozomii materni ai fătului sunt afectați. Cursul și simptomele bolilor nu au nimic în comun.
etiologia
Starea patologică se manifestă datorită disfuncției capacității de lucru a secțiunii q11-13 a celei de-a 15-a perechi de cromozomi. Sindromul Prader-Willi nu este niciodată asociat cu cromozomii materni.
Factorii care afectează evoluția bolii:
- pierderea q11-13 a cromozomului patern (acoperă 70% din cazuri);
- copia paternă a celui de-al 15-lea cromozom nu este observată, ea este modificată de copia maternă dublată (apare la 20% dintre pacienți);
- moleculele sunt transformate fără o reacție a secvenței de nucleotide ADN, care provoacă dezactivarea funcțională la făt (observată la 5% din toți pacienții).
Boala are un mod autosomal dominant de moștenire, dar un număr semnificativ de cazuri identificate sunt sporadice.
Expresia genică selectivă a tatălui și mamei este monitorizată, dar cercetătorii anterioare au presupus că transformarea informații ereditare în proteine \u200b\u200bsau ARN apare în mod egal. În zilele noastre, patogenia anomaliei rămâne neînțeleasă pe deplin.
Oamenii de știință sugerează că procesele mai lente de descompunere a grăsimilor provoacă obezitate la copii. Tironizază - o enzimă care catalizează oxidarea fenolilor, datorită muncii sale pasive, promovează hipopigmentarea pielii, părului și irisului.
Disfuncția hipotalamusului are o relație directă cu hipogonadismul manifest.
Clasificare
Există astfel de fenotipuri ale sindromului Prader-Willi:
- fenotip tipic (progresează datorită ștergerii unei copii a cromozomului tatălui);
- fenotip ușor (apare din cauza disomiei materne uniparentale);
- fenotip pronunțat (apare datorită disomiei materne uniparentale și trisomiei mozaicului 15).
Sindromul Prader-Willi conform ICD-10 are un cod unic - Q87.1
Simptome
Simptomele sindromului Prader-Willi sunt diferite, variază în funcție de categoria de vârstă a pacientului.
Starea patologică intrauterină are următoarele semne:
- scăderea activității mișcărilor fetale;
- locație neobișnuită;
Imediat după naștere, simptomele sunt următoarele:
- prezentarea în creștere a fătului;
- presiune scăzută;
- scăderea reflexului de supt;
- dificultăți de respirație.
În copilărie, un copil are următoarele condiții:
- a inhibat dezvoltarea abilităților de vorbire;
- deteriorarea dinților;
- coordonarea depresivă a mișcărilor;
- utilizare un numar mare alimente;
- creștere rapidă în greutate;
- probleme de somn;
- pubertate vine mai târziu decât de obicei;
- mic de statura;
- dezvoltarea intelectuală întârziată;
- dezvoltarea psihomotorie întârziată;
- flexibilitate excesivă.
Semne la vârsta adultă:
- infertilitate;
- nu un numar mare de păr pubian;
- obezitate;
- o tendință la diabet.
Simptome generalizate diferențe externe la adulți include:
- nas dimensiuni mari, larg;
- degete înguste;
- membre mici;
- există o hipersensibilitate a pielii;
- o cantitate mare de greutate în exces;
- fruntea este înaltă și îngustă;
- acoperirea pielii și părul de o nuanță mai deschisă în comparație cu rudele.
Există o întârziere în dezvoltarea motorie și sexuală.
La pacienții cu acest lucru proces patologic observate nivel ridicat hormonul foamei - ghrelin. Scurgând de la stomac spre glanda pituitară, se transformă pe sinteza hormonului de creștere, care provoacă comportament alimentar, prin urmare, pacienții sunt întotdeauna obezi.
Diagnostice
Sindromul Prader-Willi la copii este destul de frecvent - peste 400.000 de oameni din întreaga lume trăiesc cu acest diagnostic.
Cand diagnostic în timp util prognosticul bolii este favorabil. Un rol semnificativ îl joacă cursul bolii, care poate trece de la formă ușoară în complex - depinde de corpul uman. Sindromul are influenta negativa pe organele și sistemele copilului.
Pentru a diagnostica boala, un specialist calificat trebuie să se familiarizeze tablou clinic... În plus, se folosește și testarea genetică, ceea ce face posibilă identificarea prezenței patologiei pe cromozomul 15q11-q13, a cărui subdezvoltare este o provocare a sindromului Prader-Willi.
Diagnosticul cu test genetic confirmat la aproape fiecare pacient, este cel mai eficient pentru nou-născuți, deoarece bebelușii nu sunt capabili să se plângă de simptome.
Nașterea copiilor cu patologie poate să nu meargă foarte bine cel mai bun mod, este important să știm asta înfometarea cu oxigen iar vătămarea la naștere poate complica boala.
Lipsa de cunoștințe despre sindrom duce la faptul că boala este diagnosticată greșit. Diagnostic diferentiat se efectuează cu, deoarece este aprobat în majoritatea cazurilor în locul sindromului Prader-Willi.
Tratament
Deoarece boala aparține anomaliilor congenitale, tratamentul sindromului Prader-Willi nu este asigurat. Terapia este prescrisă pentru a se normaliza procese metabolice și eliminarea simptomelor.
În timpul terapiei pacienților, se utilizează următoarele:
- alimente dietetice, care includ alimente cu conținut scăzut de calorii, bogate în vitamine și microelemente;
- crește activitatea motorie;
- medicamente orale din;
- Clomifen, care are ca scop regenerarea secreției de gonadotropine și steroizi sexuali;
- administrarea zilnică a hormonului de creștere recombinant.
Pe stadiu timpuriu se prescrie un curs de masaj dacă pacientul are hipotonie musculară.
Este necesar să se supună examinărilor în timp util cu astfel de medici:
- neurolog;
- oftalmolog;
- endocrinolog;
- psihoterapeut.
Tratament droguri poate fi însoțită de observarea unui logoped și logoped.
Având în vedere riscul enorm de apariție a oncologiei sângelui, se recomandă consultări în timp util cu medicii.
Speranța de viață a unui pacient cu sindrom Prader-Willi depinde complet de cum se dezvoltă boala, indiferent dacă este însoțită de disfuncția sistemului cardiovascular sau a oricărui alt sistem al corpului. ÎN in termeni generali prognosticul este pozitiv, mulți pacienți trăiesc peste 60 de ani.
profilaxie
La fel de măsuri preventive cuplurile căsătorite sunt sfătuite să consulte un genetician în timpul planificării conceperii unui copil, deoarece riscul de a avea un al doilea copil cu un sindrom similar este extrem de mare.
Sindromul Prader-Willi este o anomalie genetică rară caracterizată prin oprirea genelor sau, din punct de vedere științific, acest fenomen este cauzat de o lipsă de expresie, ceea ce înseamnă transferul informațiilor ereditare de la ADN la ARN. Funcția genelor în acest caz trebuie să fie inclusă în lucrare la momentul potrivit. De exemplu, băieții nu au creșterea părului facial în copilărie, iar acest fenomen începe după debut pubertate... Această boală a fost descrisă pentru prima dată în 1956.
Sindromul Prader-Willi apare cu o frecvență de la 12.000 prunci nascuti... Această boală a fost descrisă în lucrările lor de Heinrich Willie, Andrea Prader, Andrew Ziegler, Alexis Labhart, Guido Fanzoni.
Cauzele sindromului Prader-Willi
Această boală se caracterizează prin absența sau non-exprimarea a șapte gene de la al cincisprezecelea cromozom moștenit de la tată. Sindromul Prader-Willi se observă cu modificări ale cromozomului patern, iar în cazul modificărilor cromozomului matern se observă sindromul Angelman.
În setul de gene care provoacă sindromul Prader-Willi, copia genei obținute de la tată funcționează activ, dar mama nu este. Aceasta înseamnă că, atunci când majoritatea oamenilor conțin o copie de lucru a unei gene determinate, persoanele cu sindromul Prader-Willi trăiesc fără o astfel de copie.
Simptomele sindromului Prader-Willi
Pentru această boală displazie articulațiile șoldului, hipotonie (scăderea tonusului muscular), (condițiile prealabile pentru supraalimentare apar la vârsta de 2 ani). Persoanele care suferă de sindromul Prader-Willi au scăzut coordonarea motorie și densitate scazuta oasele, sunt proprietarii picioarelor și mâinilor mici, cu o statură scurtă, predispuse la somn, alintat, au o curbură a coloanei vertebrale. Pentru astfel de oameni, saliva groasă este caracteristică (scăderea funcțiilor glandelor genitale), prezența dinților răi,. Pacienții sunt caracterizați de retard mental, precum și de dezvoltare a vorbirii, pubertate întârziată și au dificultăți în stăpânirea abilităților motorii.
Sindromul Prader-Willi și semnele acestei boli sunt marcate vizual de o punte mare a nasului, o frunte îngustă și înaltă, ochi în formă de migdale, buzele înguste. Toate aceste semne și simptome nu apar la un singur pacient. Adesea, pacienții prezintă până la cinci dintre simptomele de mai sus.
Diagnosticul sindromului Prader-Willi
Boala poate fi diagnosticată înainte de naștere. Acesta este ce mobilitate redusă fătul sau poziția adesea incorectă a copilului, precum și polihidramnios (lichid amniotic excesiv).
Sindromul Prader-Willi este adesea diagnosticat după analiza genetica... Acest test se face pe un nou-născut cu hipotonie (scăderea tonusului muscular). Uneori medicii fac greșeli și diagnostică fie miopatie. Semnele unui diagnostic al bolii după naștere includ: nașterea printr-o cezariană, prezentarea în creștere a fătului, reflex slab de supt din cauza tonului muscular slăbit al copilului, dificultăți de respirație, hipogonadism.
Copiii cu sindromul Prader-Willi sunt asemănători între ei, astfel încât un genetician cu experiență diagnostică imediat boala, fără să aștepte măcar rezultatele unui test de sânge.
Sindromul Prader-Willi și sindromul Angelman
Până în prezent, este cunoscută o altă boală, care este atribuită bolii surorii din sindromul Prader Willi. Această boală a fost numită Angelman. Se caracterizează printr-o mutație a materialului genetic matern. Rezultatele celor mai multe studii independente indică astfel de cauze ale acestei boli ca mutație la o genă. Produsul acestei gene este componenta enzimatică a tuturor sistem complex degradarea proteinelor. Această boală poartă numele pediatrului britanic Harry Angelman, care a descris-o în 1965.
Tratamentul sindromului Prader-Willi
Boala, fiind o anomalie genetică congenitală, rămâne neînțeleasă pe deplin și metodele de tratament ale acesteia nu au fost dezvoltate.
Cum este tratat sindromul Prader-Willi? Cu toate acestea, există unele măsuri terapeuticepentru a îmbunătăți calitatea vieții oamenilor. De exemplu, copiii cu hipotensiune arterială au nevoie de masaj și alte terapii speciale. Utilizarea tehnicilor speciale este demonstrată pentru a îmbunătăți dezvoltarea copilului, precum și cursuri cu un logoped și logoped. Copiii de 7, 8, 9 ani cu sindrom Prader-Willi sunt prescriși hormoni de creștere, este, de asemenea, indicat terapie cu hormoni folosind gonadotropine.
Sindromul Prader-Willi la băieți se manifestă în hipogonadism, micropenie și criptorhidie (nu prolapsul testiculelor). Într-o astfel de situație, medicii recomandă fie să aștepți ca testiculele să coboare singure, fie să recomande intervenție chirurgicală cu terapie hormonală. Pentru a corecta creșterea în greutate, este prescrisă o dietă care limitează cantitatea de carbohidrați și grăsimi. Dacă obezitatea este deja prezentă, atunci cantitatea, precum și calitatea alimentelor care sunt absorbite de pacienții cu sindromul Prader-Willi trebuie monitorizate. Pentru astfel de oameni, un pofte de lup este caracteristic. Poate o complicație a cursului bolii de apnee, caracterizată prin întârzierea respirației în timpul somnului.
Prognoza sindromului Prader-Willi
Când planificați un al doilea copil, este posibil ca acești părinți risc repetat apariția acestei boli. Depinde de mecanismul care provoacă o defecțiune genetică. Riscul este mai mic de 1% în cazurile în care primul copil are o ștergere a genelor sau o disomie partenogenetică omogenă. Riscul este de până la 50%, cu condiția ca eșecul să fie cauzat de o mutație. Riscul de până la 25% rămâne cu translocarea cromozomilor parentali. În orice caz, părinții trebuie să se supună testării genetice.
Sindromul Prader-Willi și prognosticul său mențin adesea întârzierea vorbirii dezvoltare mentală... Cercetările efectuate de Freem și Kerfs au arătat că 5% dintre pacienți au un coeficient de coeficiență chimică care corespunde nivelului mediu. Aceasta echivalează cu 85 sau mai multe puncte pe scara IQ. 27% dintre pacienți au un nivel de inteligență în pragul mediei, care este estimat la 70-85 de puncte. 34% dintre pacienți au un nivel de inteligență corespunzător la 50-70 de puncte. 27% dintre pacienți sunt la nivelul decalajului mediu și sunt estimate la 35-70 de puncte. 5% dintre pacienți au o întârziere puternică și câștigă 20-35 de puncte și mai puțin de 1% au un decalaj semnificativ. Alte studii au date care 40% dintre pacienți prezintă o inteligență moderată sau redusă.
De obicei, copiii cu sindrom Prader-Willi au o memorie vizuală bună și pe termen lung. Ei pot învăța să citească, să aibă un vocabular pasiv, bogat, dar propriul lor discurs este adesea mai rău decât înțelegerea acestuia. De asemenea, memoria auditivă, abilitățile de scriere și matematica, memoria auditivă și vizuală pe termen scurt suferă și nu respectă standardele de vârstă.
Sindromul Prader-Willi este adesea însoțit de apetit crescut, deoarece nivelul hormonului ghrelin din sânge crește la pacienți. Tipic pentru pacienți scăderea concentrației somatoliberin. Unii asociază acest lucru cu al 15-lea cromozom, care are o legătură cu hipotalamusul. Alte dovezi sugerează o scădere totalul celule, precum și celule care conțin oxitocină din nucleele paraventriculare ale hipotalamusului. Dar este interesant faptul că există dovezi care sugerează contrariul: o autopsie a deceselor cu această boală nu prezintă adesea defecte în hipotalamus.