Etiologie. Principala caracteristică biochimică a X-ALD este acumularea de acizi grași saturați neramițați în țesuturile de 24 atomi de carbon și mai mult, în special acidul hexaco zanoic (C26: 0), datorită tulburărilor decăderii lor în peroxyom. Motivul pentru aceasta este insuficiența ligoroil-koa-ligazelor peroxismice catalizând formarea derivaților KOA ai acizilor grași corespunzători. Cu X-ALD, au fost găsite mai mult de 400 de mutații genetice diferite (ABCD1) care codifică proteina membranei peroxis (ADP). În același timp, în majoritatea familiilor găsesc așa-numitele mutații private (caracteristice numai acestei familii). Gena mapată pe cromozomul X (Q28). Modul în care defectul proteinei ALDP conduce la acumularea de acizi grași cu un lanț foarte lung și manifestări ale bolii - necunoscut.
Epidemiologie. În populația generală, frecvența minimă a X-ALD la bărbați este de 1: 21000 și dacă se adaugă femei heterozigote, apoi 1-17000. Prevalența acestei boli nu depinde de cursa. Membrii aceleiași familii întâlnesc adesea fenotipuri diferite.
Patofiziologie. În microscopia electronică în citoplasma celulelor de cortex suprarenale, se găsesc macrofagele Leydega și macrofagele CNS, incluziunile stratificate caracteristice, probabil de la esterii colesterol ai unei mâini grase, cu o lungime a lanțului. Ele sunt cel mai clar vizibile în celulele pachetului de cortex suprarenale, care sunt mai întâi copleșite de lipide și apoi atrofie.
În țesutul nervos a detectat deteriorarea a două tipuri. Pentru o formă cerebrală severă a bolii la copii și forme progresive progresive la adulți, demielinizarea cu un răspuns inflamator (cluster de limfocite în spațiul perivascular) este caracterizată de cea mai pronunțată în regiunea întunecată-occipitală. Cu o formă progresivă lentă la adulți - adrenomielonevopathy - găsită, în principal axonopatia distală cu înfrângerea căilor de cefalapie lungi. Răspunsul inflamator este practic absent.
Patogeneză. Încălcarea funcției suprarenale este probabil o consecință directă a acumulării de acizi grași cu un lanț foarte lung. Cuștile zonei pachetului sunt copleșite de lipide neobișnuite. Eteriorul colesterol al acestor acizi grași sunt rezistenți la acțiunea de hidroliză indusă de ACTH, care încetinește transformarea colesterolului în steroizi activi. În plus, un exces de acid gras saturat cu 26 atomi de carbon crește vâscozitatea membranelor plasmatice, care afectează starea receptoarelor și a altor funcții celulare.
Între gradul de tulburări neurologice și natura mutației sau severitatea schimburilor biochimice (judecând după nivelul în plasmă a acizilor grași cu o lungime a lanțului), corelația este absentă. Nu există nicio legătură între severitatea încălcărilor funcțiilor glandelor suprarenale și a sistemului nervos. Severitatea simptomelor și rata de progresie a bolii depinde de intensitatea răspunsului inflamator. Această reacție poate fi mediată de citokine și să sublinieze procesul autoimun, care este într-un fel indus de excesul de acizi grași cu un lanț foarte lung. Rolul CD1 antigenului lipidic a fost stabilit. Aproximativ 50% dintre pacienți nu există un răspuns inflamator. Existența unui modificator genetic, "Configurarea" centrelor hipotalamice de termoreglare la răspunsul inflamator este asumată.
Manifestari clinice. Există cinci fenotipuri ale X-ALD. Simptomele a trei dintre ele sunt caracteristice vârstei copiilor. Cu toate fenotipurile, copilul din primii 3-4 ani se dezvoltă, de obicei, în mod normal.
În cazul formei cerebrale ale copiilor, simptomele de anunțuri, de regulă, apar la vârsta de 4 și 8 ani (cel mai devreme - la 3 ani). Cel mai adesea, hiperactivitatea este sărbătorită la începutul bolii (care este adesea luată pentru manifestarea sindromului de depreciere) și deteriorarea performanței academice școlare. Copilul distinge adesea sunetele, deși este păstrată percepția discursului de ton. Are dificultăți în a vorbi la telefon și dificultăți ca răspuns la întrebările orale. Este adesea deranjată și orientare în spațiu. Alte caracteristici inițiale includ încălcarea, ataxia, schimbarea scrisului de mână, convulsii și scânteieri. Cauzele apar aproape la toți pacienții și pot fi prima manifestare a bolii. La unii pacienți, apar simptome de creștere a presiunii intracraniene sau a tumorii unilaterale în creier. În 85% din cazuri, reacția cortizolului pe ACTH este perturbată și există o umidificare ușoară a corpului. Cu toate acestea, la majoritatea pacienților cu un astfel de fenotip, funcția suprarenală depreciată este detectată după apariția simptomelor cerebrale, care servesc drept bază pentru diagnosticare. Forma cerebrală a copiilor de anunțuri de obicei progresează rapid: spasticitatea musculară crește, apare paralizia, viziunea, zvonul, vorbirea și capacitatea de a înghiți alimentele. Există o medie de 1,9 ani între apariția primelor simptome și dezvoltarea așa-numitei stări vegetative. Speranța de viață într-un astfel de stat poate depăși 10 ani. insuficiență suprarenală. La mulți pacienți, sistemul nervos nu este afectat, în timp ce alții au tulburări neurologice mici. În multe astfel de cazuri, adrenomielonevopatia se dezvoltă mai târziu.
Termenul "AD asimptomatic" indică acele cazuri în care există caracteristici de schimburi biochimice ad, dar simptomele neurologice sau tulburările endocrine sunt absente. Aproape toți pacienții cu un defect genetic apar în cele din urmă semne clinice ale bolii, dar uneori numai după 60 de ani.
Aproximativ 50% dintre femeile heterozygice dezvoltă sindrom asemănător adrenomelonevopatiei, dar cu un curs mai puțin sever și mai târziu. Eșecul suprarenale se găsește în cazuri rare.
Adrenolatechudstrofia (sphingolipidoza, boala Addison-Schilder, Zimmerling-Creitzfeld, boala de bronz, melanomina leukeodistrofie) este o boala degenerativa determinata genetica a creierului asociat cu funcția insuficienta a glandelor suprarenale.
Cauze și mecanism de dezvoltare a bolii
Adrenoleykistrofia adrenched cu cromozomul X este o boală ereditară genetică.
Cauza bolii este o încălcare a schimbului de acizi grași, datorită deprecierii probabile a metabolismului, ceea ce duce la mutație genetică. Această boală este moștenită de atributul recesiv, pe tipul de ambreiaj cu cromozomul X.
AdrenolateCodina provoacă mutația genetică, care este situată într-un umăr lung situat în cromozomul de umăr lung X.
Acest genom este codificat o anumită enzimă care este responsabilă pentru prelucrarea structurilor de lanț lung a acizilor grași. În acest caz, există o scădere a oxidării datelor de acid, ca urmare a căreia se acumulează în substanța albă a creierului, precum și în glandele suprarenale. Acest lucru duce la un eșec în funcțiile lor. Începe procesul de demielinizare a fibrelor nervoase. Boala este moștenită de o bază recesivă, adoptată cu cromozomul X, adică există o boală a femeii. Fiind purtători ai bolii și sunt bolnavi - bărbați.
Bărbați bolnavi, începând de la o vârstă fragedă, când apar semne de insuficiență suprarenală (boala Addison).
Cea mai obișnuită formă de adrenolesophyter adrenolată cu cromozomul X este adrenomieloneeuropatia. Această patologie este găsită 1: 20.000. Această boală se caracterizează printr-o acumulare anormală de acizi grași ai unei structuri de lanț lung. De regulă, acești acizi sunt în țesuturi cerebrale, piele, păr, ochi retină, bacterii și plante,
La dezvoltarea acestei boli, numărul acestor acizi în testicule, glandele suprarenale și creierul crește la 1000 de ori. Acești acizi se înscriu în corp prin alimente, precum și sintetizate în organism pe cont propriu.
Adrenolateicodistrofia este caracteristică înfrângerii electorale, așa cum se știe că băieții cu vârsta cuprinsă între 5 și 12 ani sunt bolnavi, de regulă. În același timp, boala se termină cu un exod fatal. Corectează mai ales băieții, se manifestă în vârstă de 5-12 ani cu moartea pacientului. Adrenomiyrereyreatia este, de obicei, caracteristică adolescenților (băieți). Aceste boli sunt însoțite de demielinizarea totală cu funcție suprarenală afectată.
Imagine clinică a bolii
Această boală se caracterizează prin prezența unor simptome specifice. Simptomele adrenoleykistrofiei sunt sugerate în mod clar despre prezența tulburărilor care apar în creier și glande de secreție internă:
- euforie;
- depresie;
- încălcări ale memoriei;
- încălcări ale mersului, mișcările;
- atacuri convulsive;
- tulburări de viziune;
- schimbarea puternică a starea de spirit.
Labilitatea sistemului nervos este deosebit de caracteristică a acestei boli, când pacienții pot avansa brusc de la o dispoziție plăcută relaxată, euforică la creșterea stării oprimate și sumbre, apariția depresiunilor. Deplasarea tulburărilor și a mersului sunt însoțite, la rândul său, de fenomenul quadriplegiei, când pare a fi observată pareza de toate extremitățile sau există un ton crescut, până la apariția convulsiilor convulsive.
Din partea aparatului vizual, în plus față de reducerea acuității vizuale, pareza de mușchi vizuali, se observă o scădere a reacțiilor elevului, atrofia nervului optic. Se poate observa o pierdere parțială sau completă a vederii.
Mai târziu, atașamentul tulburărilor din sistemul nervos apare, când, pe lângă schimbările în comportament și inteligență, există simptome ale daunelor TSS și a abaterilor mentale. Performanțele școlare scade, apare Disarthria, apare o pareză spastică.
Semnele de demență sunt în creștere și se dezvoltă demență. Gradul extrem de încălcări este exprimat în dezvoltarea maramiei mentale. Aceste fenomene sunt cauzate de procesele de dezintegrare în structura creierului (Talamus, Pallipum, confidențial).

Manifestările spastice în temeiul acestei boli progresează, ca urmare a căreia se poate forma paralizia membrelor, cu pierderea completă a sensibilității acestora. Există încălcări ale vorbirii; În plus, hipogonadismul este un simptom pronunțat al adrenoleycoma, adică subdezvoltarea glandelor genitale, care au dimensiuni mici și, în consecință, au redus funcțiile. Există, de asemenea, o pigmentare a pielii crescute.
Cum este împărțită boala?
Adrenoleykistrofia (AD) există în mai multe categorii de vârstă:
- Cerebrală pentru copii.
- Cerebrală cerebrală.
- ALD cerebral la adulți.
Forma pentru copii este cea mai grea. Începutul are loc cu vârste cuprinse între 2 și 10 ani, în timp ce mortalitatea poate apărea în primii doi ani de la începutul bolii. Este posibil ca, cu un tratament de succes (operația de transplant de măduvă osoasă), poate exista o extensie a pacienților la câteva decenii.
Forma adolescentă este cea mai comună. De obicei începe la vârsta de 11 ani și mai mare. Se caracterizează printr-un curs lent și progresiv al AD.
La adulți, anunțul se dezvoltă rar la o vârstă fragedă și reamintește schizofrenia sau demența. Moartea în astfel de cazuri are loc după 3-5 ani de la începutul bolii. De asemenea, se întâmplă ca boala intra într-o stare vegetativă.
Detectarea bolii
Diagnosticul adrenoleycocodesterului este realizat de:
- RMN, creierul CT;
- analiza biochimică a sângelui și a urinei;
- studii hormonale;
- stimularea hormonilor adrenocorticotropici.
În plasma din sânge (și alte fluide biologice ale corpului), acizii grași sunt găsiți pentru structurile lungi ale lanțului în cantități crescute. Aceasta indică modificările biochimice caracteristice acestei boli.
De regulă, se observă o scădere a reacției la proces cu hormoni adrenocorticotropici, conținutul de cortizol redus în sânge. Rezoneranța magnetică Imagistica și studiile informatice ale creierului sunt cele mai exacte metode de cercetare în combinație cu metodele de laborator.
Tratamentul bolii
Tratamentul adrenolephyciei vizează restricționarea admiterii la corpul de acizi grași structuri de lanț lung și terapia hormonală de înlocuire.
Pentru a reduce manifestarea semnelor bolii, acestea sunt efectuate:
- Terapie fizică specială.
- Corecție psihologică.
- Învățarea specială pentru copiii bolnavi.
- În unele cazuri, transplantul de măduvă osoasă se efectuează dacă boala este dezvăluită în timpul timpuriu. Transplantul de măduvă osoasă este deosebit de util în cazurile de paralizie cerebrală, ca formă de manifestare a adrenolesophyterului pentru copii.
Terapia dietă include un consum echilibrat de proteine, grăsimi și carbohidrați. Consumul crescut de vitamine B1 și C este arătat. Mai ales bogat în aceste vitamine de trandafir, coacăz negru, drojdie. Creșteți utilizarea unei sări a bucătarului (până la 20 de grame pe zi), iar numărul de săruri de potasiu reduc până la 2 grame pe zi. 
De asemenea, există aditivi speciali biologic activi utilizați pentru o anumită boală (ulei Lorenzo).
Adrenoleykistrofia este o boală ereditară puternică, ceea ce duce la dezvoltarea diferitelor tulburări neurologice la copii și adulți, practic netratabile. Această boală progresează foarte repede, conducând un rezultat fatal după 1-4 ani de la începutul bolii.
Prevenirea bolii
Metodele speciale de prevenire, precum și tratamentul, în cazul adrenolestefyraismului nu există. Există doar o serie de recomandări care vizează prevenirea apariției acestei patologii genetice.
Boala neurodegenerativă cauzată de afectarea ereditară a metabolismului cu acumularea în cap și măduva spinării de metaboliți provocând distrugerea mielină. Se manifestă în principal în copilărie, cu o întârziere de dezvoltare psihomotorie, tulburări motorii, leziuni ale nervilor vizuali și auditivi, hidrocefalii, atacuri epileptice. Leukodistrofia este diagnosticată în funcție de datele de stare neurologică, anamneză, studii genetice, modelul RMN sau CT de creier, analize biochimice. Tratamentul este simptomatic. În cazul detectării precoce și a progresului lent, este posibilă transplantul de sânge din cordonul ombilical sau măduvei osoase.

General
Lakeodistrofia și-a primit numele datorită leziunii materiei albe a creierului (de la leucosul grec - alb). Există aproximativ 60 de soiuri de leucodistrofie, determinate de tipul de anomalie genică și de vârsta de manifestare a manifestărilor clinice. Împreună cu leziunile inflamatorii individuale ale sistemului nervos central (de exemplu, laceuentefalita Schilder), leucodistrofia se referă la sindromul sclerozei difuze. În același timp, înfrângerea dominantă a mielinei îl aduce mai aproape de bolile demyelinizante (scleroza multiplă, memoria RAM etc.), iar formele individuale pot fi atribuite lipidozamului.
Principalele forme de leucodistrofie includ metarmatic, supofilic, celulă globală, degenerarea lui Wang Bogart Bertrand, boala lui Alexander, versiunea spike-ului GallerWord. Cele mai frecvente sunt primele 3 tipuri de leucodistrofie. Apariția lor variază de la 0,4 la 1 caz la 100 mii de nou-născuți. O serie de forme de leucodistrofie sunt atât de rare încât doar câteva sute din observațiile lor clinice sunt descrise în literatura mondială privind neurologia. În funcție de perioada de vârstă, în care debuteile de leuceoodistrofie, fiecare din forma sa poate fi împărțită în infantilă, infantilă târziu, minor și adult.
Cauze de leucodistrofie
La inima sa, fiecare leucodistrofie are o anomalie genetică a unei anumite enzime. Apariția anomaliei și localizarea mutației genetice sunt încă instalate numai pentru cele mai întâlnite forme de patologie. În majoritatea cazurilor, leuceoodistrofia are o cale autosomal-recesivă de transmisie ereditară, dar formele sale separate pot fi moștenite cu podeaua. În plus, nu numai cazuri de mutații spontane. Defectele enzimatice deterministe genetic conduce la tulburări de schimb (mai des în metabolismul lipidic) cu definirea unui anumit metabolit în structurile nervoase și organele somatice individuale, în primul rând în ficat și rinichi.
Consecința anomaliei metabolice este distrugerea cochililor de mielina de trunchiuri nervoase și căile conductoare, moartea neuronilor cu înlocuirea pânzei de glianță în creștere. Leukeodistrofia morfologică se caracterizează prin difuzie și simetrică situată în emisferele creierului, decesele mielinei, acumularea de produse de degradare a mielinei, sporite de proliferarea GLIA. În unele versiuni nosologice, leuceowowowerphia are o imagine morfologică specifică - o colorare metarmatică sau sulfivă a produselor de degradare a mielinei, acumularea în zonele de demielinizare a celulelor globale etc.
Simptomele leucodistrofiei
În majoritatea cazurilor, leucodistrofia face debuturi în copilăria timpurie. Nou-născut, de regulă, arata sanatos. Perioadele specifice se dezvoltă în mod normal, iar apoi diferitele simptome neurologice apar treptat, caracterizate prin progresie constantă. Rata creșterii simptomelor este cea mai mare, cea mai devreme manifestă leucodistrofia. Manifestările de conducere sunt oligofrenii progresive, deprecierii, auzul, episindris, pareză spastică. Primele simptome ale leucodistrofiei pot fi ataxia, tulburările musculare-tonice (hipo- sau hipertonus, Tweet muscular), manifestări extrapiramidale, schimbă comportamentul. Apoi, există epifiganțe, manifestările bulbare, zvonurile și viziunea scade, a fost observată o scădere intelectuală cu o pierdere treptată a abilităților dobândite anterior. Tulburările senzoriale nu sunt caracteristice. În etapele ulterioare ale dezvoltării bolii, paralizia, oligofrenia pronunțată, o tulburare brută de înghițire, amavrosis, surditate sunt observate. În faza terminală, rigiditatea apreciată este de obicei observată.
Tipuri de leucodistrofie
Metakhromatic leucodistrofia În funcție de manifestare are 4 opțiuni. O debutere a versiunii Innode în primele 1-3 luni. Viața întârzierii dezvoltării și a sindromului convulsiv; Copiii nu ajung la vârsta de 1 an. Versiunea laterală laterală a leuceoodistrofiei metakhromatice începe între 1 și 3 ani, cu hipotensiune musculară și slăbiciune, ataxie, întârzieri mentale (CPR). Apoi se formează tetraplegia spastică, aphazia, sindromul Pseudobulberry. În cazuri rare, pacienții locuiesc până la 10 ani. Opțiunea juvenilă se manifestă în 4-6 ani și durează o medie de 7 ani. Adultul va face debutul în a treia decadă a vieții, uneori mai târziu, speranța de viață a pacienților de la începutul clinicii variază în decurs de 10-20 de ani.
Sudanofile Leukodistrofia Este moștenit cu un cromozom H și are mai multe soiuri. Leukodistrofia lui Peliceus Merzbacher poate începe pe primul an de viață sau în 3-4 ani. Primul semn este nistagm cu vedere mare, mai târziu există un SRR, o ataxie cerebelchiică, hipercine, pareada. Cea mai mare progresie are loc sub vârsta de 10 ani, atunci boala are o încetinire a iernii remisii. Pacienții pot trăi la vârsta matură. Adrenoleykodistrofia - o opțiune în care leukeodistrofia este combinată cu insuficiența suprarenală. Se caracterizează prin cursul progresiv cu un rezultat fatal după 6-8 ani de la începutul clinicii.
LEUKODOWOWOUTWORD-CELUAL (Boala Crabbe) - Lipoid cu acumulare în focul de demielinizare a galactocibridului și formarea celulelor globale rotunjite mari. Opțiunea anticipată se dezvoltă în prima jumătate a vieții cu hiperoportabilitate și hipertermie periodică, este întârziată dezvoltarea psihomotorie, tonul muscular este în creștere, atunci este posibilă tetrapreza spastica, oligofrenia, episindris, opistonus se dezvoltă. Într-un an, vine un rezultat fatal. Opțiunea laterală târzie este mai rară, manifestă insuficiență.
Degenerarea sprongiară a lui Wang Bogart Berran Se caracterizează printr-un episindrom, un hipermith, pronunțat de hidrocefalie cu o creștere a dimensiunii capului, ceea ce determină atrofia de amavrosi a nervilor optici. O hipertensiune ascuțită intracraniană duce la o discrepanță a cusăturilor craniene înregistrate la radiografia craniului. Pacienții cu această formă de leuceoodistrofie mor la vârsta de 3 ani.
Boala lui Alexandru (Leukodistrofia cu formare fibroasă) se datorează mutației genei responsabile pentru sinteza proteinei GFAP. Rezultatul este acumulat în celulele proteinei GFAP anormal, care conține fibre de pensionare. Opțiunea neonatală are un flux grav de rezultate fatale până la sfârșitul primului an. Opțiunea infantilă are loc la aproximativ jumătate din cazuri, se manifestă în primii 1-2 ani de viață a SRR, apoi pareză spastică, ataxie, hidrocefalie. Copiii mor câțiva ani mai târziu. Alexandru leucodistrofia juvenilă a debutelor în perioada cuprinsă între 4 și 10 ani, încasări cu simptome de stem. Speranța de viață variază în decurs de 10-30 de ani. Adultul se distinge prin manifestarea târzie și de flux relativ lent în decurs de 10 sau mai mult.
LakeodyStrofia Galerie Valp. Cel mai adesea începe la 10 ani. Se manifestă prin disfuncție a unui sistem umbros, apoi pe fundalul HyperkinesoV progresează tetrapetele, hemoralopia și retinita de pigment se dezvoltă, se observă scăderea inteligenței, apar epifiganțe.
Diagnosticarea leucodistrofiei
Căutarea de diagnosticare necesită implicarea unui număr de specialiști: neurolog, pediatru, genetică medicală, pentru a diagnostica tulburările de vedere și auzul - otolaringolog și oftalmolog. Anamneza bolii (vârsta și simptomele debutului, secvența de dezvoltare a clinicii) și istoricul familiei (prezența leucodistrofiei la rude) este importantă. Neurozonografia prin encefalografie de primăvară și ecou la pacienții mai în vârstă, de regulă, relevă o creștere a presiunii intracraniene. Leukodistrofia este însoțită de o creștere semnificativă a concentrației de proteine \u200b\u200bdatorită distrugerii celulelor cerebrale, determinată în studiul fluidului cefalorahidian.
Pentru a diagnostica tipul de anomalie metabolică, se efectuează un număr de teste biochimice cu determinarea nivelului de enzime și acumularea metaboliților. Focurile de demyelinizare este bine vizualizată cu RMN, poate fi detectată pe CT a creierului. De obicei, demielinizarea este vizibilă pe RMN al creierului înainte de manifestarea clinică a leucodistrofiei. Datorită dezvoltării geneticii, leukeodistrofia a dezvoltat diagnosticarea ADN, iar formele sale individuale (metarmatic, adrenoleycodistrofia, celula globală) este posibilitatea diagnosticului prenatal.
Tratamentul leucodistrofiei
Până în prezent, leukeodistrofia nu are modalități eficiente de tratament, permițând oprirea progresiei simptomelor. Tratamentul simptomatic este realizat - în principal deshidratare și terapie anti-wurală. Singura metodă capabilă să crească speranța de viață a pacienților cu leucodistrofie și să îmbunătățească calitatea vieții lor este transplantul de sânge bipot sau transplantul de măduvă osoasă. Transplantul duce la normalizarea metabolismului. Cu toate acestea, acest proces durează mult timp (de la 12 la 24 de luni), în timpul căruia va continua progresia leukeodistrofiei. Prin urmare, adesea grav handicap sau moartea pacientului are loc chiar și după transplantul de succes.
Trebuie subliniat că transplantul nu afectează deficitul neurologic deja dezvoltat, vă permite să vă suspendeți progresul ulterior. Datorită faptului că efectul unui astfel de tratament are loc după 1-2 ani, este recomandabil în cazul diagnosticului precoce precal al leucodistrofiei (cu vigilența corespunzătoare a părinților copilului născut din cauza prezenței unei astfel de patologii în familie) sau cu o versiune progresivă lentă a fluxului. În plus, este necesar să se țină seama de faptul că transplantul este asociat cu un risc de o serie de complicații grave, cum ar fi respingerea, reacția "transplantului împotriva gazdei", dezvoltarea infecțiilor.
AdrenoleukodyStrofie legată de X (ALD, leucododie sudanofilă, boala Schilder)
Descriere
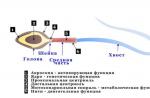
ADRENULESOPHITELOR (AD) este o boală genetică ereditară rară. Total descrise 34 tipuri de anunț. AD-ul strâns al lui X este cea mai comună formă. ALD duce la degenerare:
- Myelin Shell - acoperire de izolare a grăsimilor pe fibre nervoase din creier;
- Suprarenale și înconjurătoare cortexul lor suprarenale, care produc hormoni vitali.
Cauzele adrenoleykistrofiei
ALD este cauzată de genomul defect moștenit.
La persoanele cu AD, organismul produce acizi grași în proporțiile necorespunzătoare. Acest lucru duce la un exces de acizi grași saturați în creier și cortexul suprarenal. Ca urmare, deteriorarea cochiliei mielinei în creier și glandele suprarenale.
Factori de risc
Factorii care cresc riscul de apariție a leucodistrofiei Sudanofil includ:
- Prezența unei mișcări a unei gene defecte cu adrenolesophidie cu x-strâns;
- Vârsta: copilărie, vârstă înainte de vârsta majorității;
- Sex: bărbat, bătrânul este bolnav foarte rar.
Simptomele adrenolesophystice x-strânse
Simptomele pot varia în funcție de tipul de anunț.
AD-ul agitat (anunț cerebral pentru copii)
Acest formular este cel mai greu. Ea se găsește numai la băieți. Simptomele încep de obicei între 2-10 ani. Aproximativ 35% dintre pacienți pot avea simptome grele într-un stadiu incipient. În medie, moartea are loc în termen de doi ani. Unii pacienți pot trăi câteva decenii.
Simptomele inițiale includ:
- Schimbări comportamentale;
- Afectarea memoriei.
Pe măsură ce progresul bolii se dezvoltă mai multe simptome grave. Acestea includ:
- Pierderea vederii;
- Atacuri;
- Pierderea auzului;
- Dificultăți în înghițire și conversație;
- Dificultăți la mersul și coordonarea mișcărilor;
- Vărsături;
- Oboseală;
- Îmbunătățirea pigmentării ("bronzarea") pe piele, datorită lipsei hormonului suprarenal (boala Addison);
- Demență progresivă;
- Starea vegetativă sau moartea.
AD (AD (AD (adolescentul cerebral adolescent)
Acest tip este similar cu ALD cerebral al copiilor. Începe la 11 - 21. Și progresează, de regulă, mai lentă.
Adrenomieloneeropatie (AMN)
Aceasta este cea mai comună formă. Simptomele AMN pot apărea după 20 de ani. Boala progresează încet și poate include:
- Slăbiciune, răsturnare, pierdere în greutate, greață;
- Încălcări și depresie emoționale;
- Problemele musculare (de exemplu, probleme la mers);
- Probleme sexuale sau impotență;
- Disfuncția suprarenală;
AD-uri strânse (Adulți cerebrali de publicitate)
În același timp, simptomele sunt, de obicei, rareori manifestate la tineri (până la 20 de ani) sau vârsta medie (până la 50 de ani). Această formă de anunț cauzează simptome similare cu schizofrenia și demența. De obicei, boala progresează rapid. Moartea sau starea vegetativă poate apărea în 3-4 ani.
Formă heterozygică de ald
Acest formular se întâlnește numai la femei. Simptomele pot fi moderate sau grele. Funcționarea glandelor suprarenale, forma heterozygică a anunțului nu afectează de obicei.
Diagnosticarea adrenolesophysticului aglomerat
Când sunt suspectate de AD, sunt fabricate următoarele teste:
- Teste de sânge, pentru a determina nivelul acizilor grași;
- Creierul RMN, pentru a-și evalua starea și funcționalitatea.
Tratamentul adrenoleophicului x-strâns
Nu există niciun tratament pentru adrenolelesofencul x-strâns. Cu toate acestea, deficiențele suprarenale pot fi tratate cu cortizon. Anunțul duce, de obicei, la moarte timp de 10 ani de la apariția simptomelor.
Unele tipuri de terapie pot reduce manifestarea simptomelor de anunțuri. Uneori se efectuează tratamentul experimental.
Terapia pentru a reduce simptomele anunțului include:
- Fizioterapie;
- Terapie psihologică;
- Educație specială (pentru copii).
Unele tipuri de tratament includ:
- Transplantul de măduvă osoasă. Această operație poate fi cea mai utilă în anunțul cerebral pentru copii;
- Terapia dietetică, care include consumul:
- Produse cu conținut foarte scăzut de grăsimi;
- Uleiuri Lorenzo - suplimente nutritive bazate pe un amestec de uleiuri gliceroltrice (GTO) și GlyceratTreerukata (GTE);
- Recepția medicamentului de lovastatină - anticholessterol.
Prevenirea adrenolesophysticului
Nu există nici o modalitate de a preveni apariția anunțului. Cu condiții preliminare genetice pentru boală, este necesar să se decidă asupra nașterii copiilor.
Diagnosticul și tratamentul precoce pot preveni dezvoltarea simptomelor clinice. Acest lucru este potrivit în special pentru băieții tineri care sunt tratați cu unt Lorenzo. Noile tehnologii pot permite în curând identificarea timpurie a bolii prin screening.
Adrenoleykistrofia (AD) este un grup de boli degenerative ale SNC, care sunt adesea asociate cu insuficiență suprarenală și sunt transmise în funcție de tipul recesiv al X-legat. Clasic adrenoleykistrofia (AD) debutează la vârsta de 5-15 ani sub forma unei scăderi a performanțelor școlare, schimbărilor comportamentale și tulburărilor de mers.
Adrenoleykistrofia (AD) este cauzată de mutația în gena ABCD 1 pe cromozomul XQ28, care codifică sinteza transportatorului peroxizomic, care este implicată în transportul acizilor grași cu un lanț foarte lung în interiorul peroxisomului. Incidența adrenolateicodistrofiei (AD) este de aproximativ 1: 20.000 de băieți. În stadiile incipiente, se caracterizează convulsii generalizate.
Semne de leziune de piramide căi Include spastica și contracțiile, apare atacuri, tulburările protesute sunt exprimate datorită paraliziei Pseudobulbar - aceste simptome prevalează în etapele finale ale bolii.
Redus funcțiile glandelor suprarenale a observat aproximativ 50% din cazuri; Insuficiența suprarenală cu pigmentarea patologică a pielii (umbra întunecată, maronie fără expunere la soare) poate preceda debutul simptomelor neurologice. La demielinizarea perivativericulară CT și RMN, pornind de la secțiunile din spate, se răspândește treptat la substanța albă în departamentele din față de emisfere mari.
Auditiv VP Tver, ZVP, PSF în stadiile incipiente pot fi în normă, deoarece se dezvoltă boala, decelerarea latentălui este detectată și formularul de semnal se schimbă. Moartea are loc în termen de 10 ani de la debutul tulburărilor neurologice, dar Katamnez pe termen lung a arătat că transplantul de măduvă osoasă într-un stadiu incipient poate preveni progresia bolii.
Adreneeelopathy. Începe cu un paraperi spastic lent progresiv, incontinență urinară și impotență în deceniul 3-4 al vieții, deși semnele de insuficiență suprarenale sunt posibile din copilărie. Cazurile de adrenoleophycie tipică (AD) în familiile din familii sunt descrise în familii, în care a fost diagnosticată adrenomielonevopatia.
Una dintre dificultățile diagnosticarea x-adrenoleykistrofiei (AD) Este că pacienții dintr-o familie au observat adesea un curs clinic diferit al bolii. De exemplu, băiatul este diagnosticat cu adrenoleteicodistrofia (AD) cu un flux greu și rezultat fatal la vârsta de 10 ani, fratele acestui pacient a remarcat adrenomielonevopatia cu debutul târziu, nu au existat simptome ale bolii la al treilea frate. Consilierea genetică în familiile în care sunt dezvăluiți băieții la stadiul preimpomic al bolii, deoarece nu există metode fiabile pentru prezicerea cursului clinic al bolii.
Adrenoleykistrofia neonatală (AD) se caracterizează prin hipotensiune pronunțată, întârzierea de dezvoltare psihomotorie severă și debutul precoce al convulsiei. Tipul de moștenire este autosomal-recesiv. Lipsa fixării vizuale se datorează atrofiei nervilor vizuali. Teste care determină funcția glandelor suprarenale fără tulburări, cu toate acestea, în timpul unui studiu patologic postumus, se găsește atrofia suprarenală. Corectarea insuficienței suprarenale pentru a preveni ineficiența progresiei tulburărilor neurologice.
- reveniți la Cuprins " "