– воспалительно-аллергическое поражение мелких и средних сосудов (капилляров, венул, артериол), протекающее с образованием некротизирующих эозинофильных гранулем. Для синдрома Черджа-Стросс характерны гиперэозинофилия, поражение бронхо-легочной системы, сердца, ЖКТ, центральной и периферической нервной системы, кожи и суставов. Диагноз синдрома Черджа-Стросс основан на данных анамнеза, клинической картины, лабораторных исследований, рентгенографии органов грудной клетки, биопсии легких. В качестве основной терапии синдрома Черджа-Стросс показано назначение системных глюкокортикостероидов и цитостатиков.

Общие сведения
Синдром Черджа-Стросс – разновидность системного васкулита с гранулематозным воспалением сосудов среднего и мелкого калибра и преимущественным поражением респираторного тракта. Синдром Черджа-Стросс относится к полисистемным нарушениям, чаще всего затрагивающим органы с богатым кровоснабжением - кожу, легкие, сердце, нервную систему, ЖКТ, почки. Синдром Черджа-Стросс во многом напоминает узелковый периартериит , но в отличие от него поражает не только мелкие и средние артерии, но и капилляры, вены и венулы; характеризуется эозинофилией и гранулематозным воспалением, преимущественным поражением легких. В ревматологии синдром Черджа-Стросс встречается редко, ежегодная заболеваемость составляет 0,42 случая на 100 тыс. населения. Синдромом Черджа-Стросс страдают люди от 15 до 70 лет, средний возраст пациентов составляет 40-50 лет; у женщин заболевание выявляется несколько чаще, чем у мужчин.
Причины
Причины синдрома Черджа-Стросс неизвестны. Патогенез связан с иммунным воспалением, пролиферативно-деструктивными изменениями и повышением проницаемости сосудистой стенки, тромбообразованием, кровоизлияниями и ишемией в зоне повреждения сосудов. Важную роль в развитии синдрома Черджа-Стросс играет повышенный титр антинейтрофильных цитоплазматических антител (ANCA), антигенными мишенями которых являются ферменты нейтрофилов (главным образом, протеиназа-3 и миелопероксидаза). ANCA вызывают преждевременную дегрануляцию и нарушение трансэндотелиальной миграции активированных гранулоцитов. Сосудистые изменения приводят к появлению многочисленных эозинофильных инфильтратов в тканях и органах с образованием некротизирующих воспалительных гранулем.
На первый план при синдроме Черджа-Стросс выходит поражение легких. При гистологическом исследовании выявляются интерстициальные и периваскулярные эозинофильные инфильтраты в стенках легочных капилляров, бронхов, бронхиол и альвеол, перивазальных и перилимфатических тканях. Инфильтраты имеют разнообразную форму, обычно локализуются в нескольких сегментах легкого, но могут распространяться на всю легочную долю. Кроме острофазных воспалительных реакций, отмечаются рубцовые склеротические изменения в сосудах и легочной ткани.
Спровоцировать развитие синдрома Черджа-Стросс могут вирусная или бактериальная инфекция (например, гепатит В , стафилококковое поражение носоглотки), вакцинация , сенсибилизация организма (аллергические заболевания, лекарственная непереносимость), стрессы, охлаждение, инсоляция, беременность и роды.
Симптомы
В своем развитии синдром Черджа-Стросс проходит три стадии.
Продромальная стадия может длиться несколько лет. При типичном течении синдром Черджа-Стросс начинается с поражения респираторного тракта. Появляются аллергический ринит , симптомы носовой обструкции, полипозные разрастания слизистой носа, рецидивирующие синуситы , затяжные бронхиты с астматическим компонентом, бронхиальная астма .
Вторая стадия синдрома Черджа-Стросс характеризуется повышением уровня эозинофилов в периферической крови и тканях; проявляется тяжелыми формами бронхиальной астмы с сильными приступами кашля и экспираторного удушья, кровохарканьем. Приступы бронхоспазма сопровождаются выраженной слабостью, длительной лихорадкой, миалгией, похуданием. Хроническая эозинофильная инфильтрация легких может привести к развитию бронхоэктатической болезни , эозинофильной пневмонии , эозинофильного плеврита . При появлении плеврального выпота отмечаются боли в грудной клетке при дыхании, усиление одышки.
Третья стадия синдрома Черджа-Стросс характеризуется развитием и доминированием признаков системного васкулита с полиорганным поражением. При генерализации синдрома Черджа-Стросс степень тяжести бронхиальной астмы уменьшается. Период между появлением симптомов бронхиальной астмы и васкулита составляет в среднем 2-3 года (чем короче промежуток, тем неблагоприятнее прогноз заболевания). Отмечается высокая эозинофилия (35-85%). Со стороны сердечно-сосудистой системы возможно развитие миокардита , коронарита, констриктивного перикардита , недостаточности митрального и трехстворчатого клапанов, инфаркта миокарда , пристеночного фибропластического эндокардита Леффлера . Поражение коронарных сосудов может стать причиной внезапной смерти больных синдромом Черджа-Стросс.
Для поражения нервной системы характерны периферическая нейропатия (мононейропатия, дистальная полинейропатия «по типу перчаток или чулок»; радикулопатии , нейропатия зрительного нерва), патология ЦНС (геморрагический инсульт , эпилептические приступы , эмоциональные расстройства). Со стороны ЖКТ отмечается развитие эозинофильного гастроэнтерита (абдоминальные боли, тошнота, рвота, диарея), реже - кровотечения , перфорация желудка или кишечника, перитонит , кишечная непроходимость .
При синдроме Черджа-Стросс возникает полиморфное поражение кожи в виде болезненной геморрагической пурпуры на нижних конечностях, подкожных узелков, эритемы, крапивницы и некротических пузырьков. Часто наблюдаются полиартралгии и непрогрессирующий мигрирующий артрит . Поражение почек встречается редко, носит невыраженный характер, протекает в форме сегментарного гломерулонефрита и не сопровождается ХПН .
Диагностика
Больные синдромом Черджа-Строcс за первичной помощью обычно обращаются к различным специалистам - отоларингологу , пульмонологу, аллергологу, неврологу , кардиологу , гастроэнтерологу и поздно попадают к ревматологу . Диагностика синдрома Черджа-Стросс основана на клинико-лабораторных данных и результатах инструментальных исследований. Диагностическими критериями синдрома Черджа-Стросс считаются: гиперэозинофилия (>10% от общего числа лейкоцитов), бронхиальная астма, моно- или полинейропатия, синусит, эозинофильные инфильтраты в легких, экстраваскулярные некротизирующие гранулемы. Наличие не менее 4-х критериев подтверждает диагноз в 85% случаев.
При синдроме Черджа-Стросс также выявляется анемия, лейкоцитоз, повышение СОЭ и уровня общего IgE. Для более половины случаев синдрома Черджа-Стросс характерно обнаружение перинуклеарных антител с антимиелопероксидазной активностью (рANCA).
Рентгенография органов грудной клетки при синдроме Черджа-Стросс позволяет обнаружить быстро исчезающие, ограниченные затемнения и очаговые тени в легких, наличие плеврального выпота. При биопсии легкого определяется гранулематозное воспаление мелких сосудов, инфильтраты в околососудистом пространстве, содержащие эозинофилы. Дифференциальную диагностику синдрома Черджа-Стросс следует проводить с узелковым полиартериитом, гранулематозом Вегенера , хронической эозинофильной пневмонией, идиопатическим гиперэозинофильным синдромом, микроскопическим полиангиитом .
Лечение синдрома Черджа-Стросс
Лечение предполагает длительное назначение высоких доз системных глюкокортикостероидов. По мере улучшения состояния дозу препаратов снижают. При наличии поражений сердечно-сосудистой системы, легких, множественного мононеврита возможно применение пульс-терапии метилпреднизолоном. При неэффективности глюкокортикостероидов используются цитостатики (циклофосфамид, азатиоприн, хлорбутин), которые способствуют более быстрой ремиссии и снижению риска рецидивов, но создают высокий риск инфекционных осложнений. Перед началом терапии отменяются все лекарственные препараты, к которым у больного выявлена сенсибилизация.
Прогноз
Без лечения прогноз синдрома Черджа-Стросс неблагоприятный. При полиорганном поражении происходит быстрое прогрессирование синдрома Черджа-Стросс с высоким риском смертельного исхода от сердечно-легочных нарушений. При адекватном лечении 5-летняя выживаемость составляет 60-80%.
Этиология и встречаемость синдрома CHARGE . Синдром CHARGE (MIM №214800) - аутосомно-доминантное заболевание с многочисленными врожденными пороками развития, вызываемыми у большинства больных мутациями в гене CHD7. Предполагаемая распространенность при рождении - 1 на 3 000-12 000.
Тем не менее появление генетического тестирования может выявлять мутации гена CHD7 в атипичных случаях, что может определить более высокую встречаемость.
Патогенез синдрома CHARGE . Ген CHD7, распоженный в 8ql2, - член суперсемейства генов ДНК-связанной хромодоменной хеликазы (CHD). Считают, что белки этого семейства влияют на структурный хроматин и экспрессию генов в ходе раннего эмбрионального развития.
Ген CHD7 экспрессируется повсеместно во множестве плодных и взрослых тканей, включая глаза, улитку уха, мозг, ЦНС, желудок, кишечник, сердце, почки, легкие и печень. У пациентов с синдромом CHARGE обнаружены гетерозиготные нонсенс- и миссенс-мутации в ген CHD7, а также делеции участка 8ql2, захватывающее ген CHD7, доказывающие, что болезнь вызывает гаплонедостаточность гена.
Тем не менее некоторые пациенты с синдромом CHARGE не имеют обнаруживаемых мутаций в гене CHD7, так что иногда в основе болезни могут быть мутации в других локусах.
Фенотип и развитие синдрома CHARGE
Акроним CHARGE (С - колобома, Н - сердегные дефекты, А - атрезия хоан, R - задержка роста и развития, G - аномалии гениталий, Е - аномалии уха), охватывая наиболее частые симптомы синдрома, принят дисмор-фологами как описательное название ассоциации аномалий неизвестной этиологии и патогенеза, наблюдаемых вместе чаще, чем ожидается.
С открытием мутаций в гене CHD7 при синдроме CHARGE заболевание отнесли к дисморфическим синдромам, т.е. характерным наборам причинно связанных аномалий. Текущие основные диагностические критерии синдрома - колобома глаза (захватывающая радужку, сетчатку, сосудистую оболочку или диск, с или без микрофтальма), атрезия хоан (односторонняя или двусторонняя; стеноз или атрезия), аномалии черепных нервов (с односторонним или двусторонним лицевым параличом, нейросенсорной глухотой или проблемами глотания) и характерные аномалии слуха (наружное ухо деформировано, чашеобразное, в среднем ухе порок развития слуховых косточек, смешанная глухота и кохлеарные пороки).
Реже обнаруживают множество других аномалий , например, расщелину губы или нёба, врожденные пороки сердца, задержку роста, трахеопищеводные свищи или атрезию пищевода. Синдром CHARGE диагностируют при наличии трех-четырех специфичных критериев или двух больших и трех малых критериев.
Перинатальная или ранняя детская смертность (до 6 мес жизни), наблюдаемая приблизительно у половины больных, коррелирует с наиболее тяжелыми врожденными аномалиями, включая двустороннюю атрезию хоан и врожденные пороки сердца. Значимая причина смертности и заболеваемости - гастроэзофагеальный рефлюкс.
Часто бывают проблемы глотания ; до 50% подростков и взрослых нуждаются в установке гастростомической трубки. У большинства пациентов с синдромом CHARGE обнаруживают поведенческие аномалии (включая гиперактивность, нарушения сна и навязчивое поведение) и задержку наступления половой зрелости. Задержка физического и умственного развития может колебаться от легкой до тяжелой степени.
Поскольку исследование мутации CHD7 выявляет все больше индивидуумов с синдромом CHARGE, его симптоматика может стать более изученной, а фенотипический спектр расширится.
Особенности фенотипических проявлений синдрома CHARGE
:
Колобома радужки, сетчатки, оптического диска или зрительного нерва
Пороки сердца
Атрезия хоан
Задержка роста и развития
Аномалии полового развития
Аномалии уха
Паралич лица
Расщелины губы
Трахеопищеводные свищи
Лечение синдрома CHARGE
Если заподозрен , необходимо тщательное обследование для исключения возможной атрезии или стеноза (односторонних) хоан, врожденных пороков сердца, аномалий ЦНС, почек, утраты слуха и трудностей глотания. Оказание помощи включает хирургическую коррекцию пороков развития и тщательный уход. Важный компонент наблюдения - динамическая оценка состояния. При возможности тестирования мутаций в гене CHD7, по крайней мере, у 50% пациентов может быть установлен молекулярный диагноз.
Риски наследования синдрома CHARGE
Почти все случаи синдрома CHARGE - следствие новых доминантных мутаций с низким риском повторения у родителей. Имеется один известный пример монозиготных близнецов, имевших синдром CHARGE, а также одна семья с двумя больными сибсами (мужчина и женщина). Последняя ситуация указывает, что возможен половой мозаицизм. Если у больного обнаружена мутация в гене CHD7n оба родителя отрицательны по этой мутации, риск повторения для будущего потомства составляет менее 5%. Больной имеет 50% риск повторения у потомства.
Пример синдрома CHARGE . Девочка родилась в срок у 34-летней первоберемен-ной матери при неосложненной беременности. При родах отмечена чашеобразная форма ушной раковины справа, с поворотом ее кзади. Из-за затруднения кормления девочку перевели в отделение патологии новорожденных. Попытка проведения назогастрального зонда в правую ноздрю оказалась неудачной, что показало одностороннюю атрезию хоан. Генетик заподозрил синдром CHARGE.
Дальнейшее обследование включало эхокардиографию, обнаружившую небольшой дефект межжелудочковой перегородки, и офтальмологический осмотр, выявивший колобому сетчатки в левом глазу. Дефект межжелудочковой перегородки исправлен хирургически без осложнений.
В период новорожденности при скрининге на снижение слуха тест пройден отрицательно, и впоследствии диагностирована нейросенсорная глухота. Поиск мутаций в гене синдрома CHARGE, CHD7, показал наличие в экзоне 26 мутации 5418C>G в гетерозиготном состоянии, приводящей к образованию преждевременного стоп-кодона (Tyr1806Ter). Поиск мутации у родителей результата не дал, указывая, что мутация у ребенка произошла de novo, поэтому семью проинформировали о низком риске повторения при будущих беременностях. В возрасте 1 года девочка умеренно задержана в моторном и речевом развитии, ее рост и масса тела находятся в 5-м процентиле, окружность головки - в 10-м процентиле. Запланированы ежегодные осмотры.
Charge синдром – это редкое заболевание, возникающее во время раннего развития плода и затрагивающее несколько систем органов.
Аббревиатура происходит из первой буквы наиболее распространенных особенностей, наблюдаемых у этих детей:
- (C) = колобома (обычно ретинохороидальная) и дефекты черепных нервов (80-90%);
- (H) = сердечные дефекты (50-60%);
- (R) = замедление роста, развития (70-80%);
- (G) = недостаточная развитость половых органов из-за гипогонадотропного гипогонадизма;
- (E) = аномалии, сенсорная потеря слуха (> 90%).
Диагностика основана на определенном наборе признаков. В дополнение к особенностям, большинство детей с синдромом чардж имеют характерные черты лица:
- асимметричный паралич лицевого нерва;
- расщелина губы или неба;
- атрезия пищевода (слепая пищевая трубка);
- трахеотофагеальная фистула.
Симптомы варьируются от одного ребенка к другому. Причиной обычно является новая мутация гена CHD7, или редко, изменения в области хромосомы 8q12.2, где расположен CHD7.
Постнатальный спад роста, проблемы с глотанием очень часто связаны с дисфункциями черепных нервов. Трехмерные реконструкции МРТ-сканирований показали нарушения височной кости у более чем 85% пострадавших.
Синонимы
- Ассоциация CHARGE;
- Синдром Холла-Хиттнера;
- Колобома, атрезия хоан, замедление роста, развития, аномалии половых и мочевых путей, аномалии уха.

Charge синдром влияет на несколько систем органов, что приводит к возникновению множественных проблем при рождении. Другие характеристики могут не проявиться.
Диагноз должен быть сделан медицинским генетиком на основании наличия по крайней мере одного основного и нескольких незначительных или случайных критериев.
Основные диагностические критерии (4 C):
Coloboma – это неспособность закрыть глазное яблоко во время развития плода. Может привести к образованию зрачка в форме ствола (радужная колобома), аномалиям сетчатки, макулы или зрительного нерва.
Очень маленькие глаза (микрофтальмия), отсутствующие глаза (анофтальмия) могут быть тяжелыми формами патологии. Колобомы сетчатки или зрительного нерва приводят к значительной потере зрения, включая слепые пятна, проблемы с восприятием глубины или легальной слепотой. Она чаще всего встречаются в сетчатке и присутствует у 70-90% пациентов с синдромом CHARGE.
Двусторонние крупные ретинохороидальные колобомы являются типичной офтальмологической особенностью для людей с подтвержденными мутациями CHD7. Однако, даже глаза с большими колобоматами могут образовывать макулы.
Многие дети, имея просто радужную колобому чувствительны к яркому свету (светобоязнь). Хирургия не может исправить проблему. При близорукости или дальнозоркости помогают очки. Солнцезащитные очки и шляпа с защитным козырьком спасают от фотофобии.

Аномалии черепных нервов
Сенсинологическая (нервная) потеря слуха обусловлена отклонениями в черепном нерве VIII. Краниальная КТ часто обнаруживает гипопластическую улитку (81%) с отсутствующими полукруглыми каналами.
Потеря слуха и трудности с балансом являются наиболее распространенными особенностями, связанными с кохлеарной гипоплазией, отсутствующими полукруглыми каналами.
Синдром charge связан с характерным видом ушей, которые выступают.
Потеря слуха варьируется от легкой до глубокой глухоты. Проблемы слуха очень трудно распознать у маленьких детей. Многие дети получают кохлеарные имплантаты при сенсоневральной глухоте. У большинства есть проблемы с балансом (вестибулярные аномалии), связанные с отсутствующими полукруглыми каналами, что является ключевым показателем при постановке диагноза.
У большинства детей с чардж синдромом есть проблемы с глотанием (черепные нервы IX / X). Они включают неспособность координировать сосание и глотание, приводя к заглатыванию и аспирации пищи в легкие (может вызвать пневмонию).
Многие нуждаются в кормлении через гастростомическую трубку (прямо в желудок через брюшную стенку), пока не научатся безопасно глотать.
У многих присутствует асимметричный лицевой паралич, затронута одна сторона лица (черепно-мозговой нерв VII). Это приводит к отсутствию выражения на лице, что важно, когда ребенок работает с учителями или терапевтами.
Присутствует уменьшенное обоняние (черепный нерв I), что усложняет учебу, нормальное питание. Большинство пациентов с синдромом CHARGE имеют отсутствующие или аномальные обонятельные луковицы на МРТ, что свидетельствует об уменьшенном обонянии.
Исследование на запах может предсказать наличие гипогонадотропного гипогонадизма. Сочетание дефектного обоняния (аносмия, гипосмия) с гипогонадотропным гипогонадизмом (называемое ) приводит к небольшим внешним гениталиям. Это очень распространено при чардж и требует консультации с эндокринологом.
Changal atresia
Choanae – это место от задней части носа до горла, которое позволяет дышать через нос. Примерно у половины всех детей с расстройством эти проходы заблокированы (атрезия) или сужены (стеноз). Хирургия может исправить эти недостатки.
Пациенты с односторонней атрезией обычно корректируются 1 хирургической процедурой в более позднем возрасте (от 6 месяцев до 18 лет). С двусторонней формой нуждаются в помощи 2 вмешательств в раннем возрасте (диапазон 6 дней-6 лет).
Если обе стороны затронуты, необходимо принять немедленные меры, чтобы новорожденный мог дышать должным образом и предотвратить респираторную недостаточность.
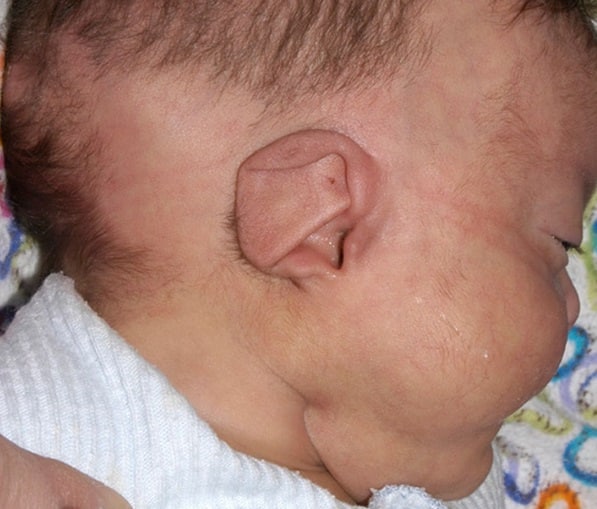
Ухо
У детей с charge синдромом необычные уши. Типичное ухо короткое, широкое с небольшой или отсутствующей мочкой. Спираль (внешняя складка) может внезапно закончиться в середине. Центр(concha) часто треугольной формы. Уши гибкие, торчат из-за слабого хряща.
CHARGE и Kabuki, являются результатом потери функциональных мутаций ДНК-связывающем белке хромодомина геликазы 7 (CHD7 ) и лизин (K) метилтрансфераза 2D (KMT2D ), соответственно. Хотя два синдрома клинически различны, существует значительное фенотипическое перекрытие.
Эпидемиология ретиноевой кислоты
Очень редкое заболевание, вызванное воздействием плода на ретиноевую кислоту (или изотретонин, который используется для лечения угрей) во время беременности. Ушные пороки похожи, однако другие функции разные.
Диагностика
Врач проводит физический осмотр на основные и второстепенные признаки расстройства, перечисленные выше. Другие подобные нарушения должны быть исключены, такие как:
- синдром делеции 22q11.2;
- расстройство Мовата –Вильсона;
- синдром Кабуки;
- синдром Каллмана;
- гаплоинтенсивность EFTUD2 (множественные врожденные аномалии, умственная неполноценность, характеризующиеся ассоциацией мандибулофациального дизостоза с нарушением уха, слуха, расщелиной неба, атрезия хонала, микроцефалия, умственная инвалидность, атрезия пищевода, врожденные пороки сердца, дефекты лучевых лучей).
CHD7 и KMT2D функционируют на тех же механизмах модификации хроматина, что дает объяснение фенотипического перекрытия между синдромами Кабуки и CHARGE.
Молекулярное генетическое тестирование доступно для мутаций CHD7. Если анализ отрицательный, следует провести SNP, поскольку иногда происходит субмикроскопическое изменение хромосомы 8q12.2. Когда оба теста отрицательны, необходимо провести секцию генома, поскольку другие ошибки имеют схожие клинические особенности.
Лечение
Хотя у этих детей много проблем, они могут выжить и стать здоровыми, счастливыми гражданами. Структурные аномалии (хелатная атрезия, сердечные дефекты, расщелина губы) корректируются хирургически.
Проблемы с кормлением и дефицит речи, требуют многих лет терапии и других вмешательств. Врачи, которые наблюдают за пострадавшими: генетики, кардиологи, аудиологи, ЛОР, офтальмологи, урологи, эндокринологи.
Более 50% испытывают нарушения сна, обструктивное апноэ во сне. Все обычные методы лечения обструктивного апноэ уменьшают симптомы.
Признание аномальных венозных структур во время отологической хирургии имеет решающее значение для предотвращения потенциально катастрофического кровотечения.
Из-за последствий дефицита кохлеарной нервной системы в принятии решений о лечении кохлеарной имплантации МРТ-оценка восьмого нерва должна рассматриваться у пациентов с тяжелой сенсоневральной потерей слуха.
Для пациентов с выраженной аномальной анатомией среднего уха, операция с использованием КТ-изображения полезна. Этим детям рекомендован двуязычный подход к раннему обучению, используя язык жестов и вербальный, чтобы обеспечить наилучшие языковые результаты. Кохлеарная имплантация дает
Обычные дозы гормона роста (GH) положительно влияют на его краткосрочную скорость без каких-либо проблем. Гормональная терапия помогает лечить симптомы гипогонадизма.
Профессионалы, участвующие в лечении, включают дефектологов, профессиональную терапию, физиотерапию, логопедию. Методы помощи и образовательные программы должны учитывать любые нарушения восприятия. Интеллект детей с charge часто недооценивается из-за общих проблем слуха и зрения.
Генетическая консультация требуется пострадавшим людям и их семьям. Другое лечение является симптоматическим и поддерживающим. Для этих сложных детей необходим многодисциплинарный медицинский подход.

Особенности развития, прогноз
Большинство маленьких детей с чардж синдромом имеют задержки психофизического развития. Это происходит прежде всего из-за сенсорного дефицита, частых заболеваний, госпитализаций.
Многие догонят сверстников в более позднем детстве, проявляя нормальные интеллектуальные способности. Невозможно предсказать возможное развитие. Раннее вмешательство сурдопедагога имеет важное значение для устранения сенсорного дефицита, предотвращения поведенческих проблем.
Независимо от степени внутренних аномалий уха и интеллектуальных способностей, по мере взросления могут возникнуть сложности поведения.
Синдром Чардж / Charge(СЧС) - врожденное заболевание, характеризующееся врожденными патологиями развития различных органов. Развивается либо в результате генетической мутации (мутация гена CHD7), либо под воздействием внешних факторов.
Вы путаете синдром charge и синдром чардж-стросса.
Аббревиатура CHARGE
- С- coloboma (колобома);
- H - heard defect (патология сердца);
- A-atresia choanae (атрезия хоан);
- R- retarded growth and development (задержка роста и развития);
- G - genital abnormality - патология гениталий;
- E - ear abnormality - патология уха;
Классификационные критерии
Это 6 основных проявлений: астма, эозинофилия > 10%, моно или полинейропатия, летучие легочные инфильтраты,синусит, экстраваскулярная тканевая эозинофилия (American College of Rheumatology, 1990). Если у больного выявляются четыре из указанных шести признаков, то диагностическая чувствительность превышает 85%, специфичность 99,7%. Центральное место занимает бронхиальная астма, которая позволяет врачу ориентироваться среди других проявлений системных васкулитов.
Морфология
Патологические изменения в легочной ткани исследованы недостаточно.
Cottin и Cordier
приводят немногочисленные данные по патологическим изменениям в легочной паренхиме. Эти изменения носят распространенный и вариабельный характер; наиболее выраженными из них являются некротические изменения и образование каверн. Во многих сосудах выявляются тромбы и участки кровоизлияний, на более поздних стадиях обнаруживают разрастание рубцовой соединительной ткани. Гистологические изменения при СЧС характеризуются сочетанием некротизирующейся гранулемы, васкулита мелких и средних сосудов, а также развитием эозинофильной пневмонии. У больных, которых не лечили стероидными препаратами, выявляются обширные эозинофильные инфильтраты, преимущественно интерстициальные и периваскулярные.
Некротизирующаяся воспалительная гранулема расположена экстраваскулярно, в этот патологический процесс сосуды вовлекаются редко. Гранулема характеризуется появлением некротической зоны, которая окружена эпителиодными гистиоцитами. Для этого типа гранулем типично значительное содержание эозинофилов и кристаллов ШаркоЛейдена. В пестрой морфологической картине наблюдаются также саркоидподобные гранулемы. Другим определяющим признаком первичного системного васкулита при СЧС являются морфологические изменения в стенках сосудов. В процесс вовлекаются мелкие артерии и вены, стенки сосудов инфильтрированы клетками, дифференциальнодиагностическое значение имеет появление эозинофилов и гигантских клеток. Воспалительная реакция находится на различных этапах своего развития, поэтому, помимо острофазовых реакций, наблюдаются их исходы в виде рубцовых склеротических изменений в сосудах и легочной ткани. Морфологическая картина дополняется изменениями со стороны бронхов и бронхиол, которые характерны для бронхиальной астмы. Стенка бронхов инфильтрирована эозинофилами, слизистая отечна, гладкие мышцы находятся в состоянии гипертрофии, налицо метаплазия бокаловидных клеток, происходит значительное утолщение базальной мембраны, формируются слизистые пробки в просвете терминального отдела дыхательных путей. Интерстициальная ткань легких, так же как и интеральвеолярное пространство, инфильтрирована лимфоцитами, плазматическими клетками и гистиоцитами. Морфологическая картина дополняется изменениями со стороны бронхов и бронхиол, которые характерны для бронхиальной астмы. Стенка бронхов инфильтрирована эозинофилами, слизистая отечна, гладкие мышцы находятся в состоянии гипертрофии, налицо метаплазия бокаловидных клеток, происходит значительное утолщение базальной мембраны, формируются слизистые пробки в просвете терминального отдела дыхательных путей. Интерстициальная ткань легких, так же как и интеральвеолярное пространство, инфильтрирована лимфоцитами, плазматическими клетками и гистиоцитами. Трансбронхиальная биопсия обычно позволяет получить достаточный материал для проведения гистологического исследования, и только в редких случаях рекомендуется проведение открытой биопсии легких. Типичными морфологическими чертами васкулита является выраженная инфильтрация эозинофилами стенки мелких сосудов. Важный признак первичного системного васкулита обнаружение некротизирующей гранулемы. Эти изменения могут быть обнаружены при исследовании кожи и подкожной клетчатки.
Дифференциальная диагностика
СЧС проводится с гранулематозом Вегенера, гиперэозинофильным синдромом,узелковым полиартериитом,микроскопическим полиангиитом; она не представляет трудностей, если брать за основу клинические проявления первичного системного васкулита. Однако морфологическое различие представляет определенные трудности при разграничении близких по своим проявлениям васкулитов. Наибольшую диагностическую значимость имеют некротизирующий васкулит, эозинофильная пневмония, экстраваскулярный гранулематоз, которые патогномоничны для СЧС. Так, при гранулематозе Вегенера не происходит интенсивной инфильтрации эозинофилами, в то время как образование асептической некротической полости более характерно для ранних его стадий, а при СЧС возможно лишь на далеко зашедших стадиях болезни. Экстраваскулярная гранулема не встречается при узелковом полиартериите, и поражение легких не является ведущим проявлением при этом васкулите. Более сложна дифференциальная диагностика между хронической эозинофильной пневмонией и СЧС, так как инфильтрация легких эозинофилами морфологически очень близка. Задача усложняется также и тем, что при хронической эозинофильной пневмонии могут быть обнаружены проявления умеренно выраженного васкулита. Однако некротизирующий гранулематоз встречается только при СЧС.
Клиническая картина
описали три фазы клинического течения СЧС. На естественное течение болезни могут оказывать влияние многие факторы, особенно медикаментозная терапия.
Первая фаза
.В типичных случаях болезнь начинается с проявлений аллергического ринита, который часто осложняется полипозными разрастаниями слизистой носа и присоединением синуситов и бронхиальной астмы. Первая фаза заболевания может продолжаться несколько лет, и основным клиническим синдромом является бронхиальная астма.
Вторая фаза
характеризуется повышенным содержанием эозинофилов в периферической крови и выраженной их миграцией в ткани. На этом этапе формируется хроническая эозинофильная инфильтрация легких и желудочнокишечного тракта.
Третья фаза
заболевания характеризуется частыми и тяжело протекающими приступами бронхиальной астмы и появлением признаков системного васкулита. Временной интервал между возникновением симптомов бронхиальной астмы и васкулита составляет в среднем три года (в литературе описан случай, когда он составил 50 лет). Считается, что чем короче этот интервал, тем неблагоприятней прогноз течения СЧС. Болезнь может проявиться в любом возрасте, но чаще признаки системного васкулита приходятся на четвертую или пятую декаду жизни. Женщины болеют в три раза чаще, чем мужчины. По данным эпидемиологических исследований, в клинической практике чаще встречаются больные с гранулематозом Вегенера, нежели больные с СЧС.
CHARGE syndrome is a disorder that affects many areas of the body. CHARGE is an abbreviation for several of the features common in the disorder: coloboma , heart defects, atresia choanae (also known as choanal atresia), growth retardation, genital abnormalities, and ear abnormalities. The pattern of malformations varies among individuals with this disorder, and the multiple health problems can be life-threatening in infancy. Affected individuals usually have several major characteristics or a combination of major and minor characteristics.
The major characteristics of CHARGE syndrome are common in this disorder and occur less frequently in other disorders. Most individuals with CHARGE syndrome have a gap or hole in one of the structures of the eye (coloboma ), which forms during early development. A coloboma may be present in one or both eyes and may impair a person"s vision, depending on its size and location. Some affected individuals also have abnormally small or underdeveloped eyes (microphthalmia). In many people with CHARGE syndrome , one or both nasal passages are narrowed (choanal stenosis) or completely blocked (choanal atresia), which can cause difficulty breathing. Affected individuals frequently have






